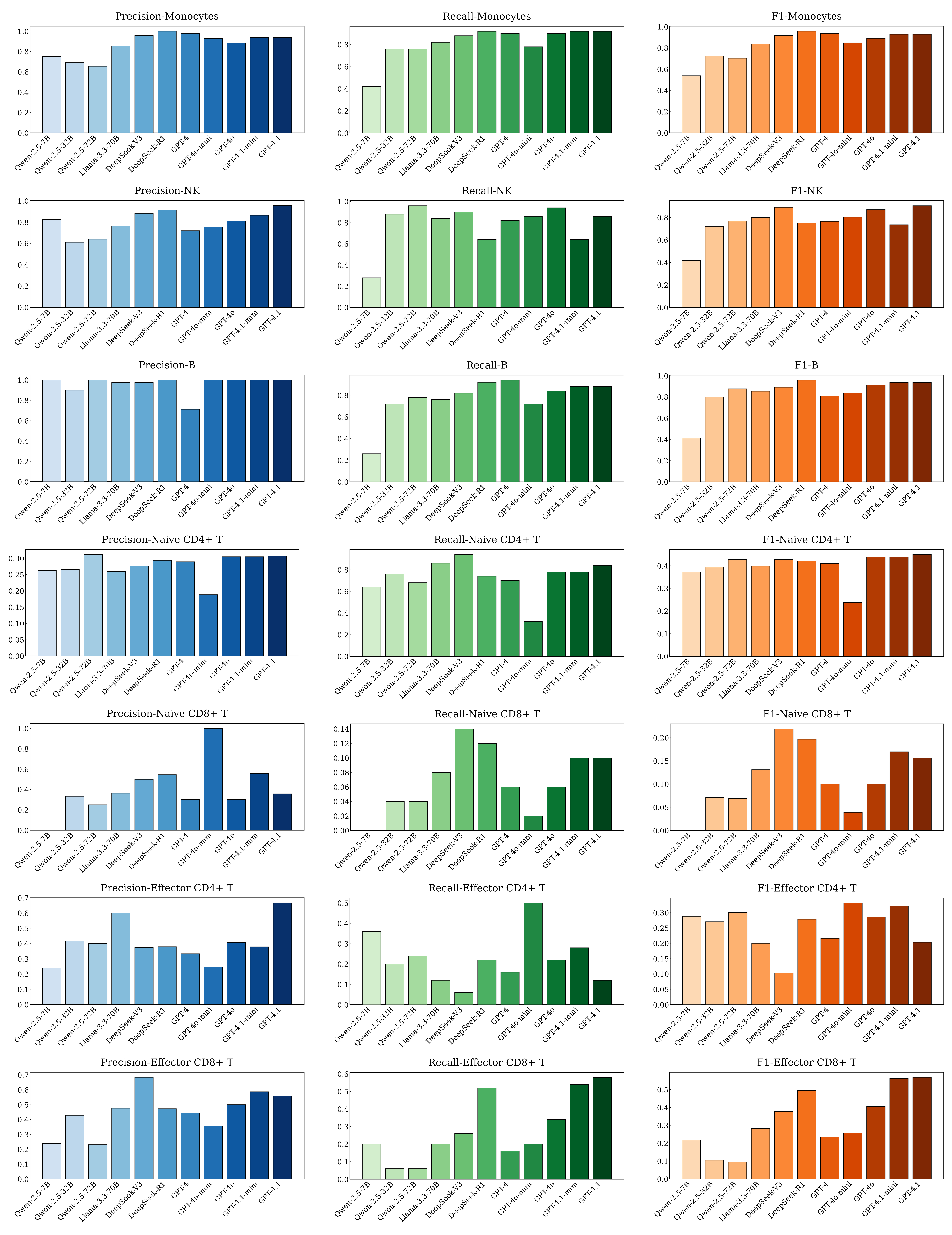
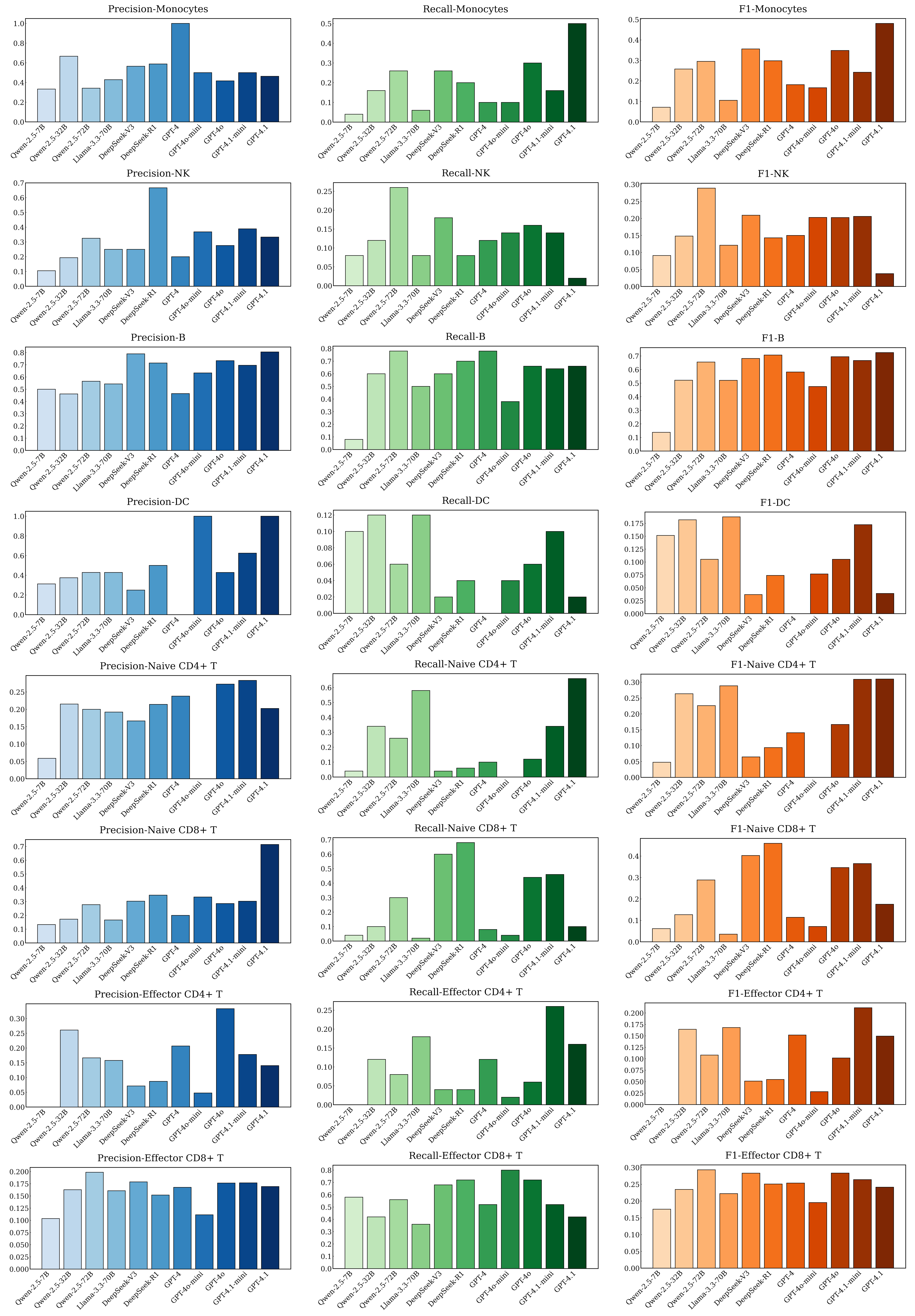
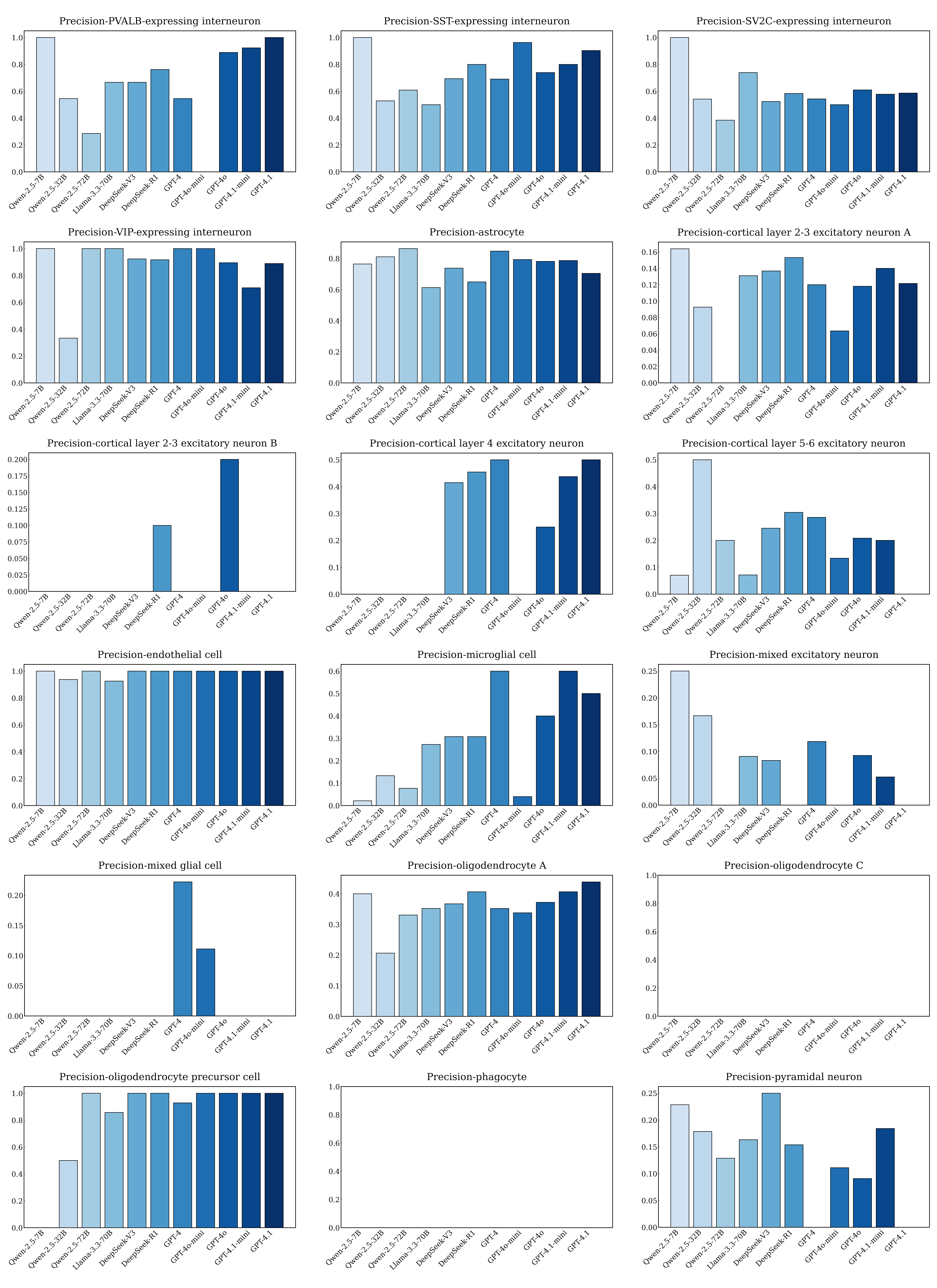
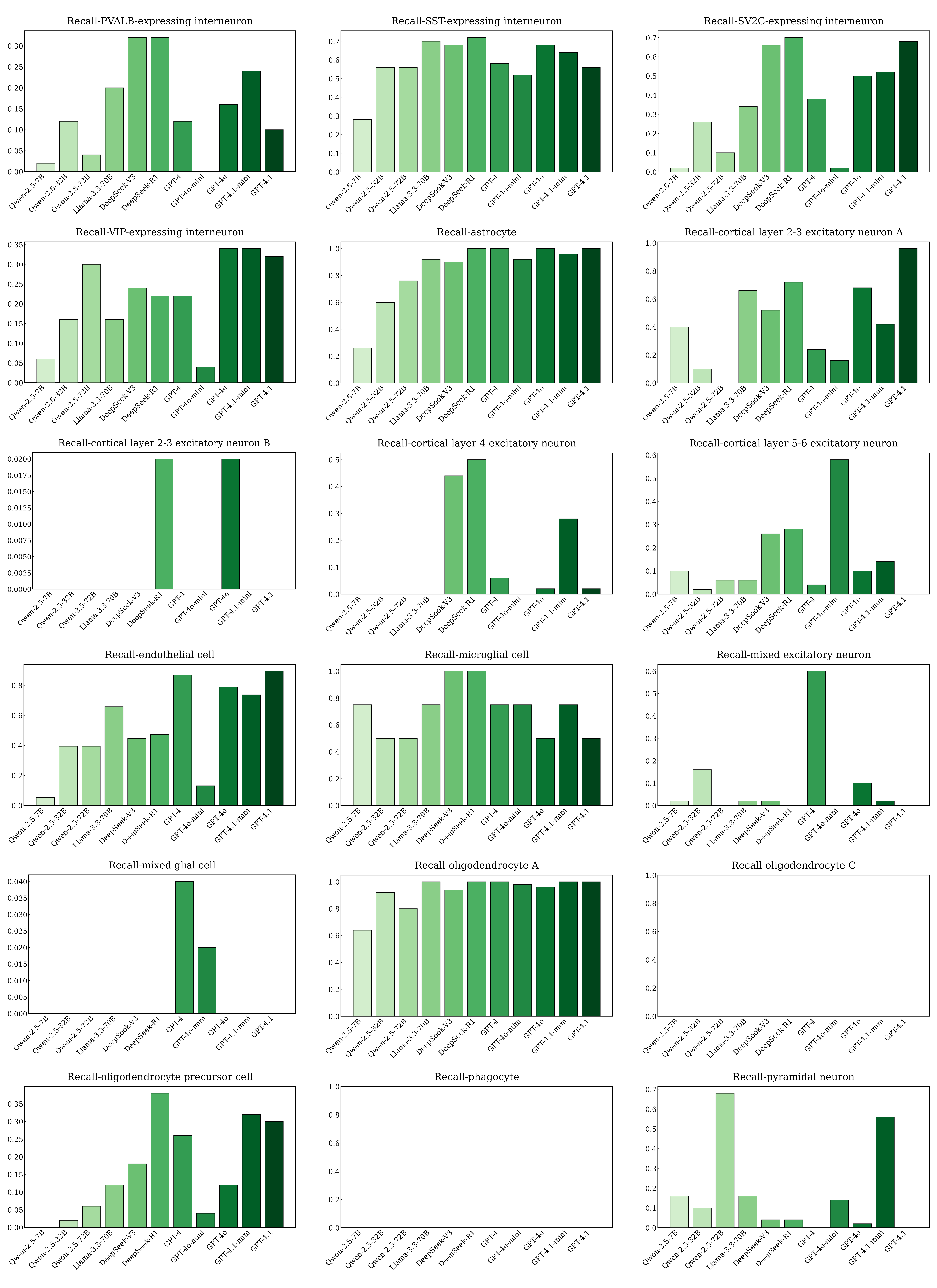
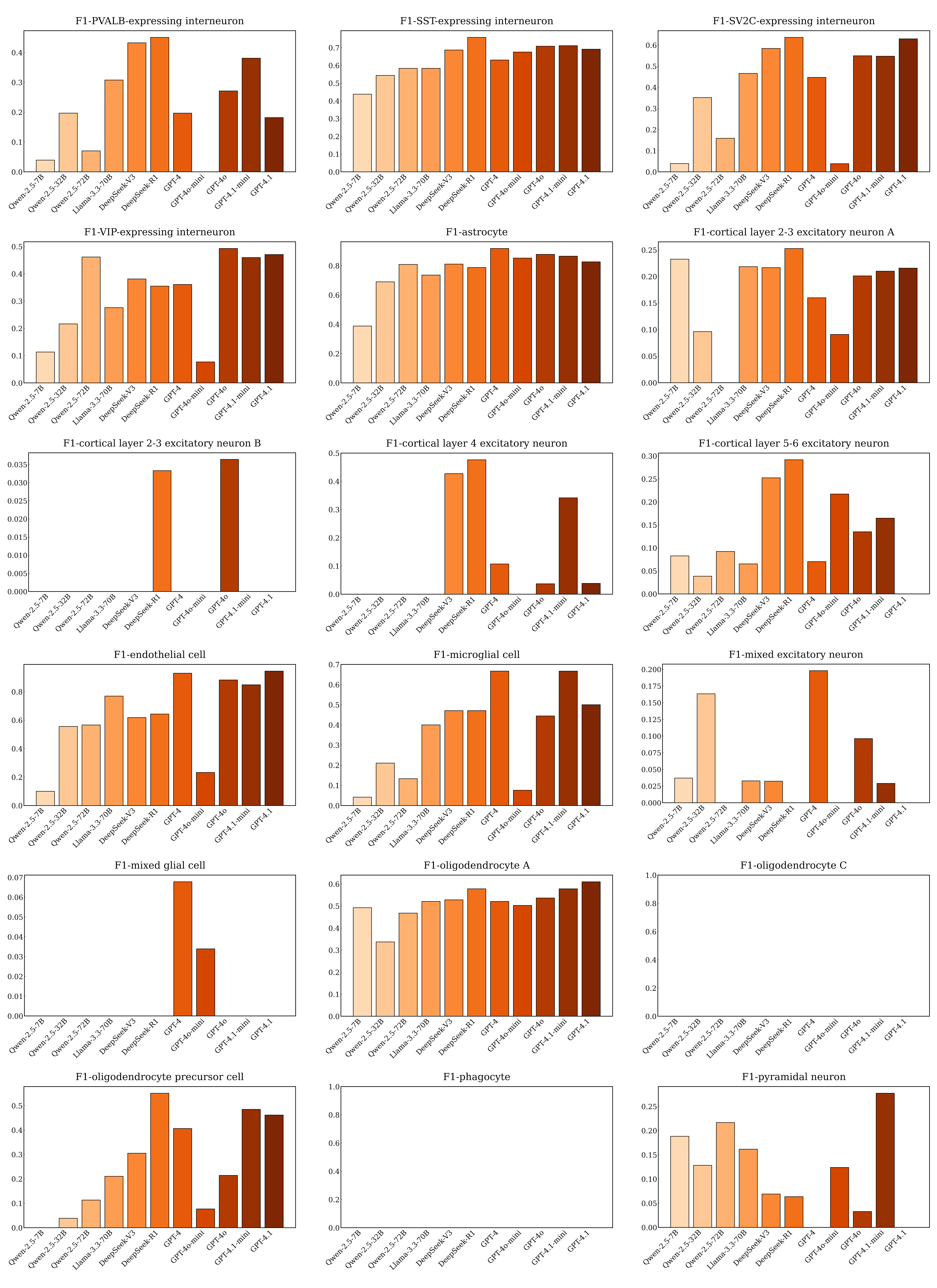
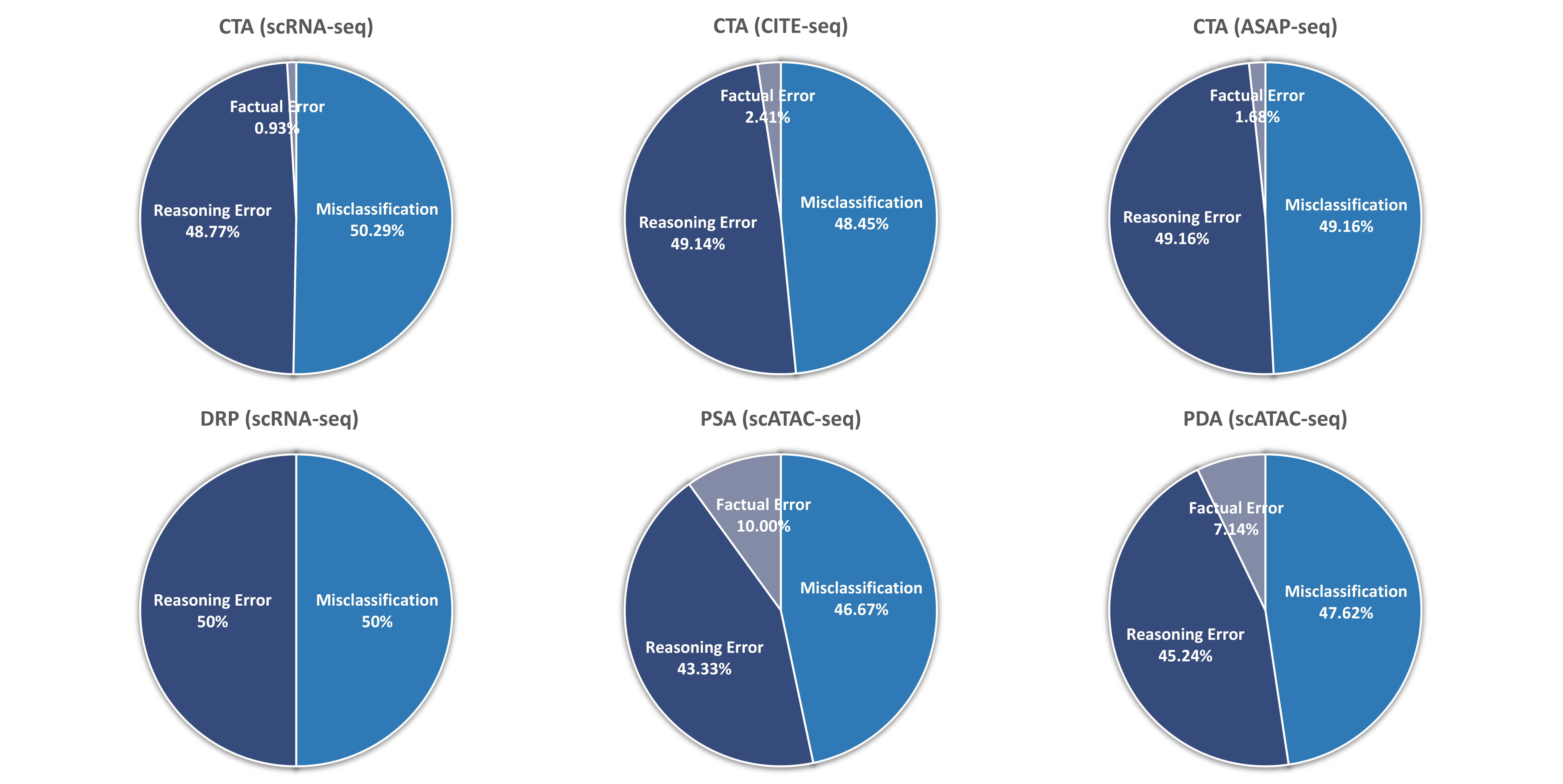
| # | Model | CTA (scRNA-seq) | CTA (CITE-seq) | CTA (ASAP-seq) | DRP | PSA | PDA | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Accuracy | F1 Score | Accuracy | F1 Score | Accuracy | F1 Score | Accuracy | F1 Score | Accuracy | F1 Score | Accuracy | F1 Score | ||
| 1 | DeepSeek-R1 🥇 | 42.38 | 33.40 | 58.29 | 50.76 | 28.00 | 20.81 | 50.00 | 33.33 | 76.67 | 58.00 | 62.96 | 57.07 |
| 2 | GPT-4.1-mini 🥈 | 40.51 | 34.55 | 59.14 | 58.04 | 29.33 | 27.47 | 55.00 | 48.19 | 68.33 | 27.61 | 61.11 | 41.63 |
| 3 | GPT-4o 🥉 | 35.70 | 29.03 | 58.29 | 55.76 | 28.00 | 25.00 | 47.50 | 22.96 | 71.67 | 61.35 | 55.56 | 54.56 |
| 4 | GPT-4.1 | 37.83 | 30.94 | 61.43 | 59.26 | 28.22 | 23.99 | 49.38 | 35.12 | 73.33 | 55.47 | 46.30 | 46.28 |
| 5 | LLaMA-3.3-70B | 32.75 | 24.99 | 52.57 | 50.02 | 22.00 | 19.30 | 43.75 | 43.67 | 66.67 | 58.96 | 57.41 | 55.56 |
| 6 | DeepSeek-V3 | 37.57 | 30.60 | 57.14 | 54.65 | 27.11 | 21.18 | 50.63 | 39.34 | 76.67 | 49.52 | 24.07 | 22.94 |
| 7 | Qwen-2.5-72B | 24.73 | 19.32 | 50.29 | 40.50 | 28.44 | 22.61 | 50.00 | 30.84 | 73.33 | 42.31 | 35.19 | 29.17 |
| 8 | Qwen-2.5-32B | 22.46 | 18.76 | 48.86 | 38.57 | 23.11 | 19.87 | 45.63 | 43.50 | 76.67 | 43.40 | 11.11 | 11.11 |
| 9 | Qwen-2.5-7B | 13.77 | 11.53 | 30.86 | 28.08 | 10.67 | 7.37 | 49.38 | 32.38 | 76.67 | 43.40 | 29.63 | 24.36 |
| 10 | GPT-4o-mini | 23.93 | 15.77 | 48.57 | 47.89 | 16.89 | 13.52 | 43.75 | 35.24 | 41.67 | 41.52 | 31.48 | 20.43 |
| 11 | GPT-4 | 35.16 | 29.89 | 53.43 | 43.44 | 21.56 | 17.16 | 1.25 | 1.57 | 0.00 | 0.00 | 0.00 | 0.00 |
| 12 | C2S-Pythia | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 |
Introduction
With the rapid progress in artificial intelligence and deep learning, methodological advances in single-cell analysis have undergone a notable shift from traditional statistical techniques to specialized deep learning models, and more recently, to pre-trained foundation models. While these developments have led to significant improvements in performance and scalability, several inherent limitations remain unresolved: (1) Lack of Unification. For different types of omics data and downstream tasks, existing paradigms typically require separately designed models, lacking a unified approach capable of simultaneously handling multi-omics and multi-task scenarios. (2) Limited User-Friendliness. Effective application of these methods to single-cell analysis often necessitates domain expertise in biology as well as proficiency in programming. Furthermore, the lack of user-centric interaction design in current models poses a significant barrier to adoption by non-expert users. (3) Poor Interpretability. Most of the existing data-driven black-box models directly learn the mapping from input (e.g., gene expression) to output (e.g., cell type information), without incorporating interpretable intermediate steps. As a result, users are often unable to understand the rationale behind the model's decisions. To this end, we seek to establish a unified, user-friendly, and interpretable paradigm for single-cell analysis.
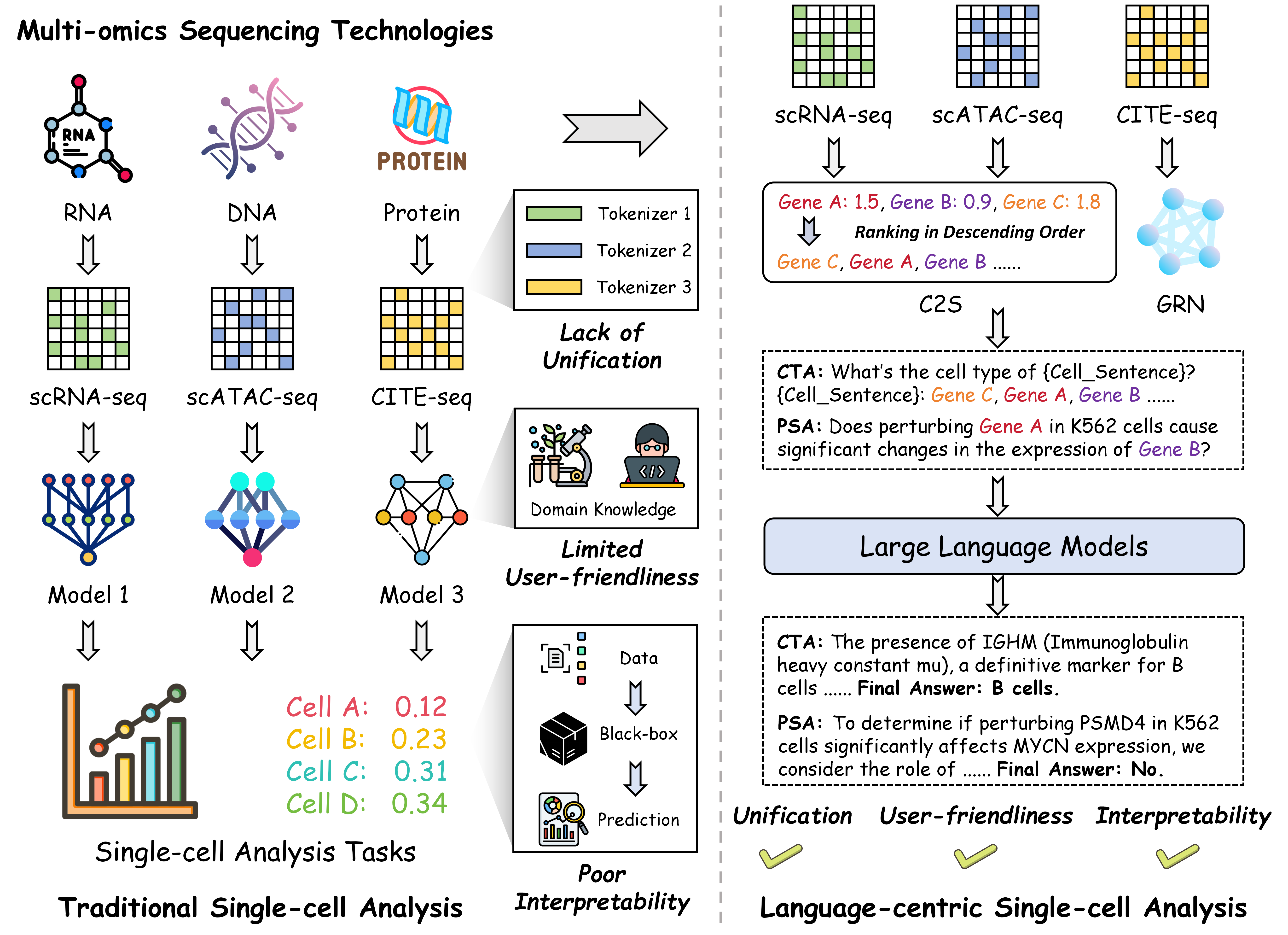
An illustration of traditional single-cell analysis and language-centric single-cell analysis.
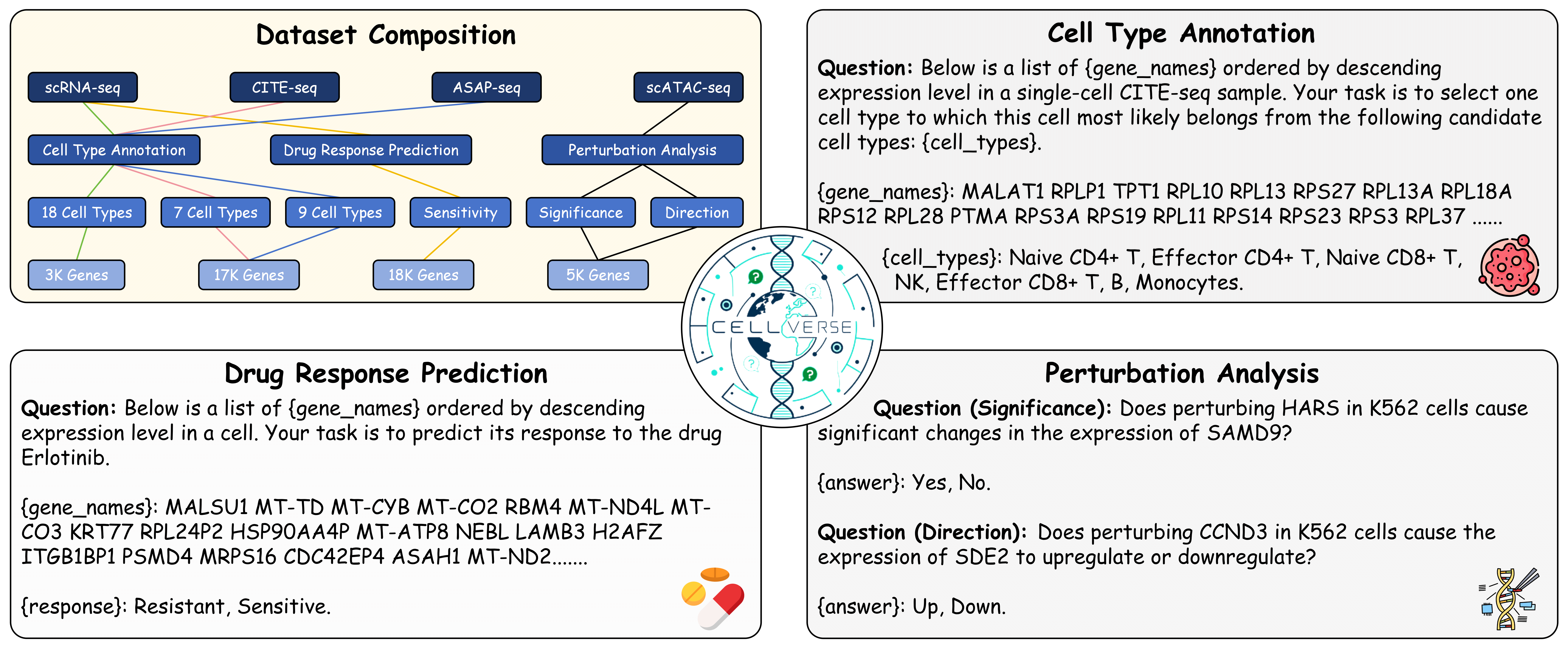
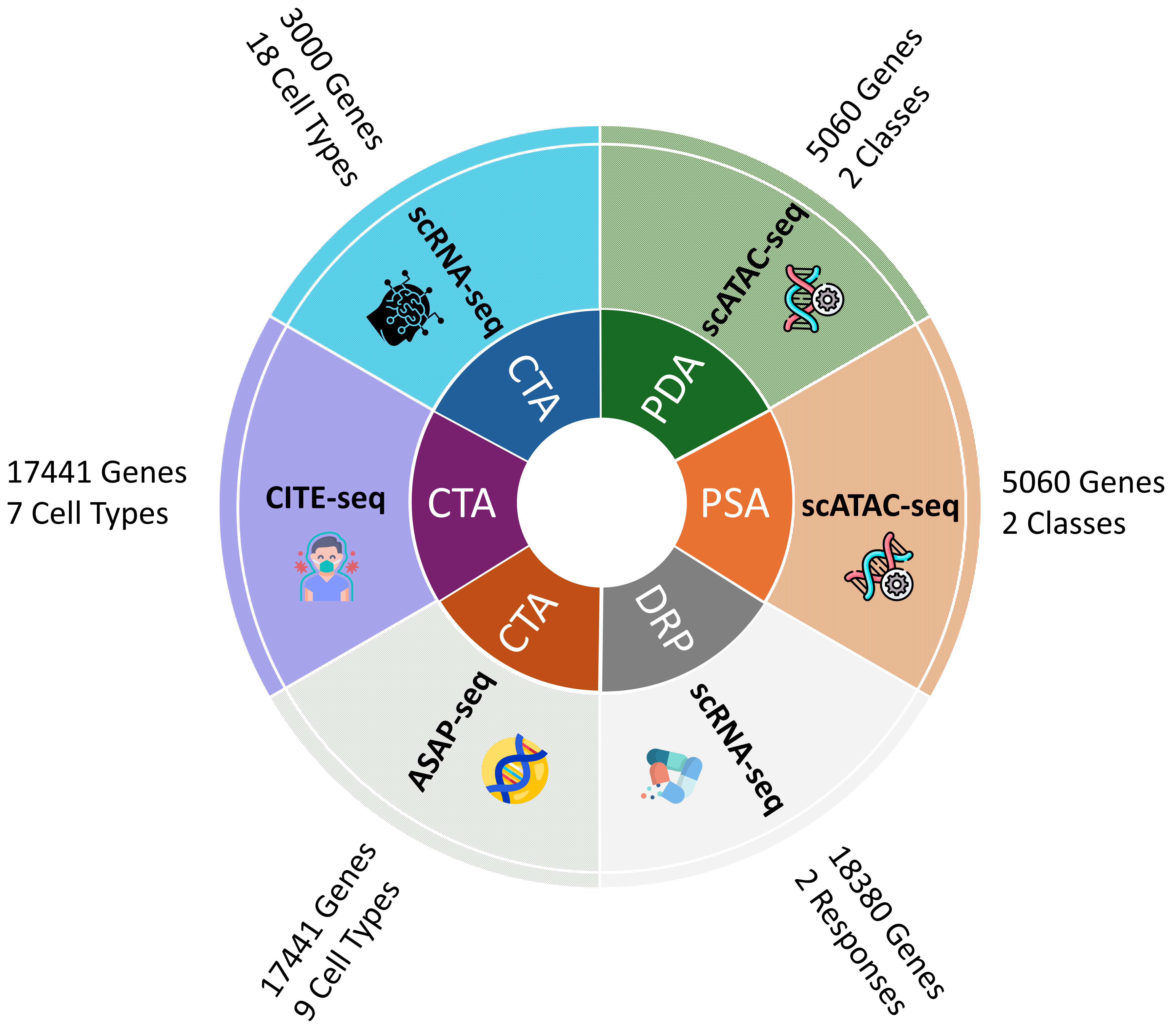
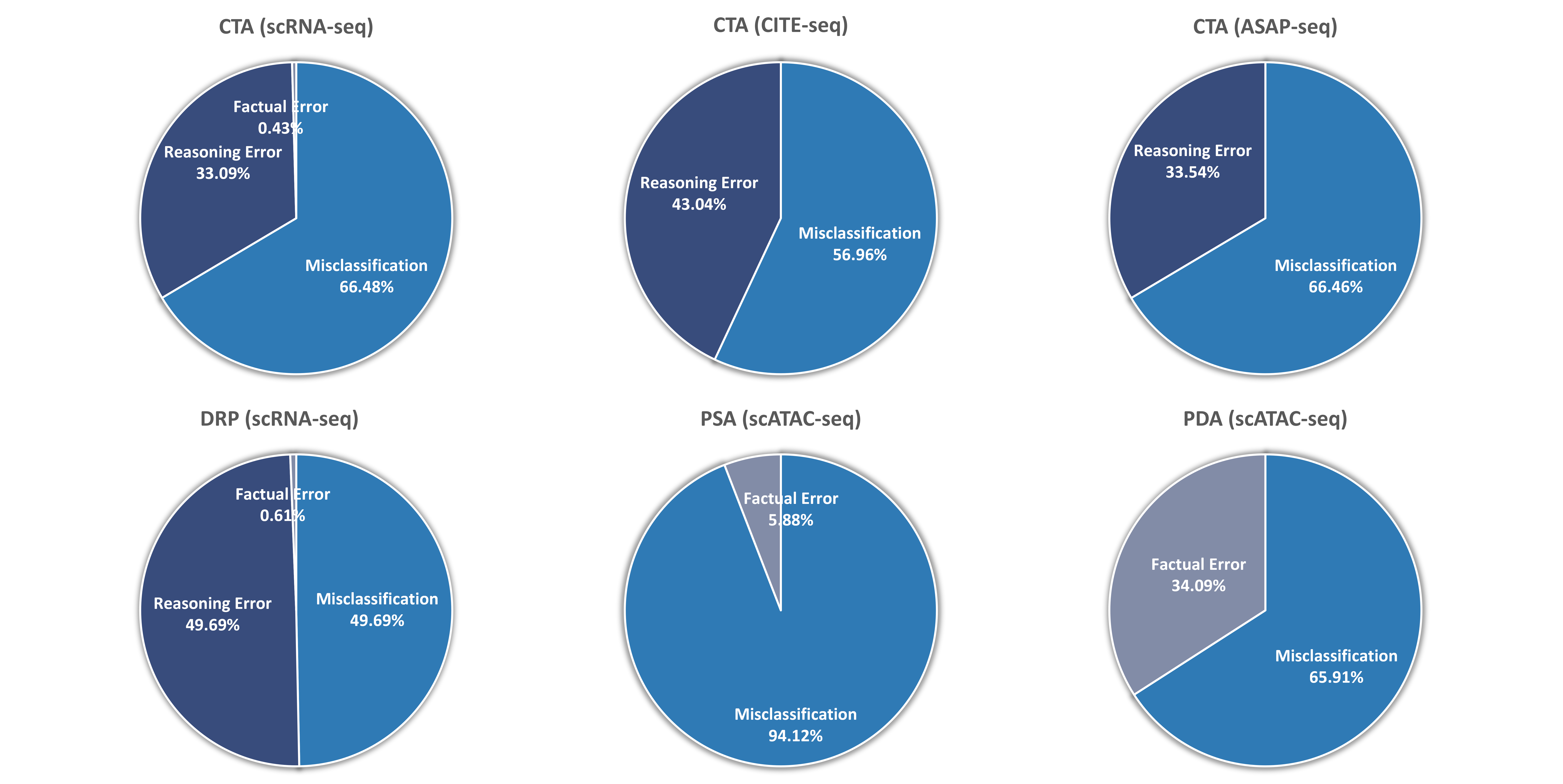
Specifically, we introduce CellVerse , a unified language-centric benchmark dataset for evaluating the capabilities of LLMs in single-cell analysis. We begin by curating five sub-datasets spanning four types of single-cell multi-omics data (scRNA-seq, CITE-seq, ASAP-seq, and scATAC-seq data) and translate them into natural languages. Subsequently, we select three most representative single-cell analysis tasks—cell type annotation (cell-level), drug response prediction (drug-level), and perturbation analysis (gene-level)—and reformulate them as question-answering (QA) problems by integrating each with the natural language-formatted single-cell data. Next, we conduct a comprehensive and systematic evaluation of 14 advanced LLMs on CellVerse . The evaluated models include open-source LLMs such as C2S-Pythia (160M, 410M, and 1B), Qwen-2.5 (7B, 32B, and 72B),Llama-3.3-70B, and DeepSeek (V3 and R1), as well as closed-source models including GPT-4, GPT-4o-mini, GPT-4o, GPT-4.1-mini, and GPT-4.1.
Through this large-scale empirical assessment, we uncover several key insights: (1) Generalist models perform better than specialist models, with DeepSeek-family and GPT-family models demonstrating emergent reasoning capabilities in cell biology, while C2S-Pythia exhibit complete failure across all sub-tasks. (2) Model performance positively correlates with parameter scale, as evidenced by the Qwen-2.5 series where the performance hierarchy follows model size: 72B > 32B > 7B variants. (3) Current LLMs demonstrate limited understanding of cell biology, particularly in drug response prediction and perturbation analysis tasks where most LLMs fail to surpass random guessing baselines with statistical significance.